扩张型心肌病 (DCM) 一词定义了一组异质性心脏疾病,其特征是左心室或双心室扩张和收缩功能障碍,但没有异常负荷条件或冠状动脉疾病足以引起收缩功能障碍。在大约三分之一的病例中,DCM是家族性的,具有遗传发病机制。尽管传统上心电图 (ECG) 在 DCM 中被认为是非特异性的,但最近获得的基因型-表型相关性知识提供了新的机会来识别可能指向特定 DCM 亚型的模式和异常。学习心电图解释并适当使用其他基于心电图的技术,包括动态心电图监测,运动耐量试验和成像方式,如超声心动图和心血管磁共振,可以早期识别特定的遗传性或后天性扩张型心肌病。此外,心电图异常可以反映疾病的严重程度,并为风险分层和管理提供有用的工具。在本文中,我们会了解ECG在DCM诊断和管理中的当前作用。我们描述了各种临床环境,在这些环境中,正确使用和解释 ECG 可以提供宝贵的线索,随着心血管医学的发展,有助于这一基本工具发挥重要作用。心电图异常可以反映疾病的严重程度,并为风险分层和管理提供有用的工具。在本综述中,我们讨论了 ECG 在 DCM 诊断和管理中的当前作用。我们描述了各种临床环境,在这些环境中,正确使用和解释 ECG 可以提供宝贵的线索,随着心血管医学的发展,有助于这一基本工具发挥重要作用。
引入
扩张型心肌病 (DCM) 目前的定义是在没有足以引起全身收缩损伤的异常负荷条件或冠状动脉疾病的情况下,存在左心室 (LV) 或双心室扩张和收缩功能障碍。扩张型心肌病是一个涵盖性定义,包括多种疾病,其中心肌异常与冠心病或瓣膜病或先天性心脏病无关,并且不能用异常的血液动力学状况来解释。除了非常通用之外,DCM一词可能不完全准确,因为腔室扩张通常不存在:事实上,“扩张”一词最近受到质疑。这种疾病分类学的复杂性建立在显著的遗传异质性之上,在至少 50 个不同的个体基因中发现与该疾病相关的突变,以及以心律失常和心力衰竭为最常见表现的多态性临床表现。
心血管成像的最新技术进步,特别是心血管磁共振 (CMR) 的日益广泛使用,为通过组织表征进行深层表型和病因学定义提供了机会,从而可以识别水肿、纤维化或浸润等。然而,在这个充满快速发展的诊断技术的复杂环境中,心电图 (ECG) 在 DCM 患者的评估中仍然发挥着极其强大的作用,它可以提供诊断危险信号,有助于确定诊断工作的后续阶段,预后分层标准和可以指导适当决策制定的信息。
在这篇文章中,我们将讨论心电图仍然是扩张型心肌病诊断、风险分层和管理中最重要难题的几个原因。在概述涉及心电图每个部分的异常之后,详细讨论了以特定模式为特征的个别疾病。
方法
心电图解释的系统方法
心电图异常是大多数 DCM 患者的特征,超过 80% 的病例报告有异常心电图特征。尽管传统观点认为 DCM 中的 ECG 异常是非特异性的,但与肥厚型心肌病 (HCM) 或致心律失常性右心室心肌病 (ARVC) 等其他心肌病相比,基因型-表型相关性的最新进展提供了有机会识别特定的 ECG 模式,这些模式是某些遗传性或后天性 DCM 的典型特征。由于DCM中的ECG很少正常,因此 ECG 异常应触发诊断检查的启动。然而,在解释心肌病患者的 ECG 时,方法应“以心肌病为导向”,即放弃源自缺血性和高血压性心脏病世界的经典概念,并关注特定的“危险信号”应仔细整合到更广泛的临床和家庭环境中。
P波
DCM 时心房经常扩张,反映出充盈压升高和/或相关的瓣膜异常。这可能反映在 ECG 上,P波变化提示左心房和/或心房扩大。虽然孤立的右心房扩大并不常见,但左心房扩大在不同比例的患者中可见,并且通常被认为是长期存在疾病的标志。心房颤动 (AF) 是所有形式的扩张型心肌病发展为心力衰竭后的常见途径。然而,年轻人早发 AF 可能提示特定的 DCM 病因,主要是遗传。
PR间期
一级和/或晚期房室 (AV) 传导阻滞可见于扩张型心肌病患者;传导异常,尤其是在年轻患者中,表明通常与神经肌肉疾病、离子通道障碍相关的特定遗传背景。传导异常在心脏结节病和南美锥虫病等后天性疾病中也相对常见。
QRS波群
重要心肌的丧失和弥漫性 LV 纤维化都可能导致 QRS 振幅降低,尤其是在心前区导联。低 QRS 电压也可能反映脂肪浸润,例如由于桥粒基因突变导致的致心律失常性心肌病,涉及左心室和右心室。当 DCM 患者符合LV 肥厚( LVH) 电压标准(Sokolow-Lyon 或 Cornell)时,应排除高血压病因。
左束支传导阻滞 (LBBB) 存在于大约三分之一的 DCM 患者中,有时先于结构表型,并且具有不良预后价值;LBBB 可能是希氏束内离散病变的结果,在远端希氏束起搏可以改善 LBBB 患者的电末端超声心动图去同步化。许多诊断为LBBB的患者同时存在LVH和左前分支阻滞,而不是真正的 LBBB。如果 QRS 持续时间≥140 ms(女性为 130 ms),V1-V2 有 QS 或 rS 模式,并且 V1、V2、V5、V6、V6、V2、V5、V6、V6、V2、V5、V6、V6、V2、V2、V6、V2、V2、V2、V6、V2、V2、V2、V2、V2、V2、V2、V2、V2、V2、V2、V2、V2、V2、QRS 导联中≥2条或QRS波中部切迹或连线出现V2导联QRS中段切迹或连贯,aVL。心脏再同步化治疗 (CRT) 后,与其他心室延迟相比,这种形态与更好的超声心动图反应相关。
右束支传导阻滞 (RBBB) 在 DCM 患者中通常不常见 (2–6%),但由于抗肌萎缩蛋白基因的致病变异,它在神经肌肉疾病患者中很常见。
Q波
关于病理性Q波的定义缺乏共识,提出了多项诊断标准,构成了混淆的根源。Q波持续时间 ≥ 40 ms 或绝对深度 > 3 mm 被一些人认为是病理学标准,而其他人则建议振幅≥随后 R 波的 25%。在没有缺血性心脏病的 DCM 中可以观察到20个Q波,并且在前壁和侧壁导联中更常见。如下所述,肌营养不良症的心脏受累通常以后部或下部 Q 波为特征,这反映了透壁心肌纤维化。29
ST段/T波异常
复极化异常在 DCM 中很常见,通常反映了 LV 损伤。T 波倒置 (TWI),尤其是在侧导联中,是某些遗传形式(例如细丝蛋白 C 或桥粒病)的公认特征。与 HCM 相比,明显的复极化异常(例如深 TWI,尤其是侧导联)很常见,而 DCM 中的 TWI 深度较浅,并且与 LVH 的电压标准无关。
QT间期
DCM中的 QTc 间期通常是正常的。短QT间期与原发性肉碱缺乏症有关,后者可能导致扩张型心肌病。长期监测的 QT 变异性已被证明可用于DCM患者的心源性猝死 (SCD) 风险分层。
室性早搏
多达40%的扩张型心肌病患者可发现室性早搏 (VPB)。频繁的 VPB 可能会促进 LV 收缩功能障碍,在某些情况下,确定VPB是否是LV收缩功能障碍(心动过速性心肌病)的主要驱动力因素可能不太容易。对于被认为足以引起 LV 收缩功能障碍的 VPB 负荷,目前还没有达成共识;然而,高负荷被定义为从 >10 000 到 25 000 VPB/天,以及在 24 小时内 >10% 到 24% 的总心跳。VPB 的类型而不仅仅是 VPB 的负担与不同形式的 DCM 的鉴别诊断相关。此外,某些 VPB 形态在运动员中很常见,被认为是良性的。这些包括漏斗状和束状形态。相反,VPB 的其他形态,如 LBBB/中间或上轴或 RBBB/中间或上轴和宽 QRS 可能是潜在心肌疾病的征兆。
遗传性 DCM 的“致心律失常”子集,以核纤层蛋白 A/C 和桥粒形式的 DCM 为代表,其特征是在疾病早期出现复杂和多形性室性心律失常(包括频繁的 VPB),预示着 SCD 的风险增加。VPB 和/或非持续性室性心动过速 (NSVT) 的存在通常并不决定在初级预防中选择使用植入式心律转复除颤器 (ICD) 来保护具有 DCM 表型的患者。然而,频繁心律失常的存在,尤其是与桥粒基因致病变异和/或 CMR 心肌纤维化相关时,可能提示 SCD 的高风险,因此在这种情况下应考虑 ICD。
运动耐量试验中室性心律失常的发展,包括 VBPs 的增加或运动期间 NSVT 的发展,可能是具有潜在桥粒致病性变异的致心律失常表型的标志。
室上性心律失常
通过动态监测识别AF是DCM管理的一个重要方面,并且可能决定重要的选择,如开始抗凝治疗以预防卒中。在年轻的 DCM 患者中检测阵发性室上性心律失常应提示对家族性 LMNA 心肌病进行调查。
总而言之,从实践的角度来看,当接诊患有无法解释的 LV 扩张和/或收缩功能障碍的患者时,从 P 波开始到 T 波结束的 ECG 系统分析可能会提供宝贵的线索,这些线索可能指向特定亚型的诊断对管理和预后有影响。
扩张型心肌病特定遗传形式的心电图
某些 ECG 特征是特定遗传性 DCM 亚型的线索,因为某些致病基因与特征性 ECG 异常相关可能对患者及其亲属具有诊断和预后价值。例如,某些基因(核纤层蛋白 A/C、细丝蛋白 C、桥粒基因和受磷蛋白)的致病变异可能表现出致心律失常表型,这应该导致在一级预防中早期决定植入 ICD。在接下来的几行中,描述了一些最常见的 DCM 相关基因型中的 ECG 模式。
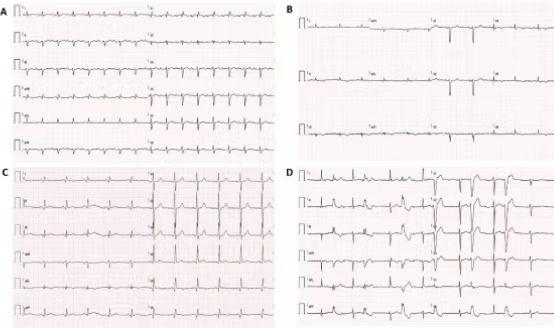
图(1)
本站所注明来源为"爱爱医"的文章,版权归作者与本站共同所有,非经授权不得转载。
本站所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,如果您认为我们的转载侵犯了您的权益,请及时通过电话(400-626-9910)或邮箱(zlzs@120.net)通知我们,我们将第一时间处理,感谢。