摘要:肝豆状核变性是一种常染色体隐性遗传的铜代谢障碍疾病。致病基因为ATP7B,其突变导致ATP酶功能减弱或丧失,使血清铜蓝蛋白合成减少以及胆道排铜障碍,导致蓄积于体内的铜离子在肝、脑、肾、角膜等处沉积[1],引起相应的临床表现,包括神经精神症状、肝生化异常、角膜K-F环、肾损害、溶血性贫血、 骨骼肌肉损害等表现。临床上分为肝型、脑型、其他类型及混合型[1]。在世界范围内WD的发病率约1/10万~ 1/3万[3],好发年龄在5 ~ 35岁[4],40 岁以后发病患者约占3%[5]。WD的临床症状可不典型,因此对肝功能异常不能用常见肝病解释的患者应及时行K-F环、血清铜蓝蛋白、24 h 尿铜、腹部超声、头颅MRI等检查协助诊断[6]。在此我们报告一例以肝病为首发表现的成人肝豆状核变性,为提高对本病的认识,减少误诊和漏诊。
案例展示:
患者女性,50岁,主因“间断腹泻、发现肝脏占位性病变2年余”于2017年6月5日就诊。腹部B超示:肝脏弥漫性病变伴多发结节(较大者直径约1. 1 cm),脾肿大。完善腹部CT提示肝左叶及脾脏形态饱满。经药物治疗后腹泻好转,查肠镜未见异常。期间未再出现不适,未规律体检。既往“肾病综合征”病史10余年,未行肾穿刺活组织检查。银屑病10 余年,潜伏梅毒10年。哥哥患肝炎去世、妹妹患肝炎。
体格检查:体温36. 6℃ ,脉博62 次/min,呼吸20次/min,血压126/76 mm Hg,BMI 27 kg/m2,皮肤黝黑,全身皮肤黏膜及巩膜无黄染,无肝掌、蜘蛛痣。全身浅表淋巴结未触及肿大。腹部微隆起,腹软,无压痛、反跳痛及肌紧张,未及包块,Murphy征阴性,肝脏肋下未触及,脾脏触诊不满意,肝区无叩痛,腹部叩诊鼓音,移动性浊音阴性。肠鸣音4次/min。双下肢无水肿。心、肺、神经系统查体未见异常。入院后完善相关检查,血常规:WBC 3. 63×109/L,Hb 146 g/L,PLT 86×109/L。尿便常规未见异常。
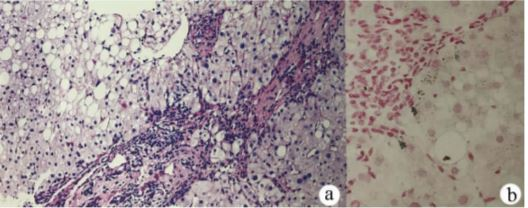
辅助检查:血生化:ALT 45 U/L,AST 36 U/L,Alb 46.3 g/L,ALP 76 U/L,GGT 36 U/L,TBil 17. 1μmol/L,DBil 2. 96 μmol/L, SCr 94 μmol/L,TG 1. 67 mmol/L,TC 5. 19 mmol/L,LDL 2. 71mmol/L。凝血:PTA 86%。乙型肝炎、丙型肝炎标志物均阴性。巨细胞病毒-IgM <5. 00 U/ml、巨细胞病毒-IgG 83. 60U/ml、EB病毒-IgM< 10. 00 U/ml、EB病毒-IgG 172. 00 U/ml。梅毒螺旋体明胶凝集试验阳性,梅毒螺旋体抗体阳性,梅毒快速血浆反应素试验弱阳性;后梅毒快速血浆反应素试验滴度1∶1。自身抗体谱:抗核抗体(ANA)阳性(颗粒型)1∶ 320,余未见异常。IgG、IgA、IgM正常、甲状腺功能未见异常。血清铜蓝蛋白23. 7mg/L;尿铜138. 5μg/24 h。角膜K - F环阴性。腹部B超(2017年6月6日):肝脏实质回声增粗,不均质,肝内占位;脂肪肝;脾肿大。FibroScan:受控衰减参数321 dB/ m,E 8. 1 kPa。肝穿刺病理:重度脂肪肝,肝内少量铜沉积,符合WD(图1)。
诊断:间断腹泻、发现肝脏占位性病变2年余。B超、CT影像学均提示肝豆状核变性征像。结合临床表现、影像学评估及术中表现确诊为"以肝病为首发表现的成人肝豆状核变性"。
鉴别诊断:应重点鉴别其他原因导致的肝脏损害。对于以神经精神症状为主要表现的患者,应重点排除具有相似临床表现的其他神经系统疾病。(1)肝硬化:肝功能异常或肝硬化,多种病因可引起肝功能异常或肝硬化,包括病毒性肝炎、酒精滥用、自身免疫性肝炎、药物导致的肝毒性、遗传性血色病和a1抗胰蛋白酶缺乏症等。对于伴随有肝外症状如神经系统症状的患者,需要高度警惕肝豆状核变性。对于不伴其他肝外症状的患者,也宜常规筛查除外肝豆状核变性的可能性。(2)小舞蹈病:神经系统疾病,锥体外系症状是肝豆状核变性的典型临床表现之一,需要与特发性震颤、风湿舞蹈病、特发性扭转性肌张力障碍,泛酸激酶依赖型神经退行性疾病等以锥体外系症状为突出表现的其他神经系统疾病相鉴别。
治疗及转归:患者于2017年6月20日开始加用青霉胺治疗,250 mg/d起始,第3天加量500mg/d;6月27日复查24 h尿铜1027. 5μg,同时对症支持治疗,驱铜治疗期间无不适,病情好转后出院。
讨论:
本例患者血清学化验结果及肝穿刺病理结果满足2008年《肝豆状核变性的诊断与治疗指南》诊断标准[1],考虑WD诊断明确。虽然没有发现典型的K-F环,但研究表明以肝损伤为主而无神经系统损害时K-F环检出率仅为44%~62%。阳性家族史对诊断WD有重要意义,对新发现WD患者的亲属尤其是一级亲属应作WD的相关项目筛查,并进行基因检测[1]。本例患者兄妹均患有肝炎,不除外家族史可能,但由于经济原因,患者本人及妹妹均未能行基因检测。回顾本例患者病史,患者2年余前行腹部超声提示肝内弥漫性病变伴多发结节,因为肝生化正常,未进一步明确病因。虽然对这种少见遗传病的认识不断增加,但是诊治延误并不少见。分析本例患者延误诊断原因:患者近2年查肝生化指标正常,而影像学异常未能引起重视。WD影像学不典型,多表现为肝实质光点回声增粗、增多、增强。但是梁娜等[6]研究认为无论以何种疾病为主要表现的WD患者,甚至无临床表现,肝脏声像图均已表现出异常。因此出现不能用普通常见疾病解释的影像学改变时,需要考虑少见疾病。目前无证据证明患者为自身免疫性肝病合并WD。目前针对WD治疗主要有2大类药物,一是络合剂,能强力促进体内铜离子排出,如青霉胺、二巯丙磺酸钠等;二是阻止肠道对外源性铜的吸收,如锌剂、四硫钼酸盐[1]。WD一经确诊需终身治疗。青霉胺是第一个用于治疗WD药物,经大量研究证实疗效确切。一般以肝病为主要表现的患者多在用药后2 ~ 6个月肝功能改善明显。维持治疗1年以上,病情趋于稳定[5]。明确诊断的患者需定期随访,监测临床或生化指标上的改善,以确保其治疗的依从性,并及时发现药物治疗的不良反应。WD是少数能有效控制的遗传病之一,若能早期诊断,大部分患者预后良好。
总结:
总之,当患者出现以下表现时应考虑本病的可能:原因不明的急、慢性肝病;年龄在7~8岁以上,出现以锥体外系为主的神经系统症状;Coomb’s试验阴性的急性血管内溶血;不明原因的血尿、肾小管功能不全;不明原因的骨关节症状。特别是伴有神经系统或精神症状的肝病患者,或者是有一级亲属患肝豆状核变性的患者,尤其要重点考虑本病可能性。明确诊断的患者需定期随访,监测临床或生化指标上的改善,以确保其治疗的依从性,并及时发现药物治疗的不良反应。 WD是少数能有效控制的遗传病之一,若能早期诊断,大部分患者预后良好。未来有望制定更加科学的有效的诊断及治疗临床指南。
参考文献:
[1]Parkinson′s Disease and Movement Disorders Study Group.Neurology Branch of Chinese Medical Association. Guidelines for the diagnosis and treatment of hepatolenticular degeneration[J]Chin J Neurol, 2008, 41(8):566 -569. (inChinese)中华医学会神经病学分会帕金森病及运动障碍学组. 肝豆状核变性的诊断与治疗指南[J]. 中华神经科杂志, 2008, 41(8):566 -569.
[2]SHAH D. Wilson′s disease: hepatic manifestations[ J]. DisMon, 2014, 60(9): 465 -474.
[3]SAMAR H, HARRIS VKN, SAMEER S. Wilson′s disease[J].Lancet, 2007, 369(9565): 902 -903.
[4]ROBERTS EA, SCHILSKY ML. A practice guideline on Wilson disease ( vol 37, pg 1475, 2003 )[ J ]. Hepatology,2003, 38(2): 536.
[5]European Association for Study of Liver. EASL Clinical Practice Guidelines:Wilson′s disease [ J]. J Hepatol, 2012, 56(3): 671 -685.
[6]ROBERTS EA, SCHILSKY ML. Diagnosis and treatment of Wilson disease:an update [J]. Hepatology,2008,47(6):2089 -2111.
本站所注明来源为"爱爱医"的文章,版权归作者与本站共同所有,非经授权不得转载。
本站所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,如果您认为我们的转载侵犯了您的权益,请及时通过电话(400-626-9910)或邮箱(zlzs@120.net)通知我们,我们将第一时间处理,感谢。